Relational DataBase, 관계형 데이터베이스
- RDB는 table과 같은 형태로 데이터를 저장하는 database를 말한다.
- 행렬, 엑셀과 같은 형태를 상상하면 쉽다.
Basic Term
Row(or record)
- 각 행은 하나의 데이터 record를 의미한다.
Column(or field)
- 각 열은 데이터의 속성을 의미한다.
- 데이터의 속성 뿐 아니라 데이터의 조건사항(schema)를 적어줄 수도 있다.
Primary Key
- 데이터들을 행 별로 구별할 때 사용할 열.
- 우리가 일반적으로 엑셀에서 table을 만들 때 가장 첫번째 열의 값들이 행을 구별할 때 사용된다는 것을 생각하자.
- Primary Key는 단 하나만 지정되어야 한다.
Foreign Key
- Primary Key를 참조하는 그 외의 열들.
- Primary Key가 데이터의 이름이라면 Foreign Key는 그 속성일 것이다.
RDB의 장점
논리적인 데이터 조직화 및 관리
CREATE TABLE customers(
id INT NOT NULL ATUO_INCREMENT,
name VARCHAR(255) NOT NULL,
email VARCHAR(255) NOT NULL,
phone_number VARCHAR(255) NOT NULL,
PRIMARY KEY (id)
);
- 위와 같은 SQL 명령어로 생성한 테이블의 경우, 고객의 ID, 이름, 이메일, 전화번호를 저장한다.
- 모든 속성들을 하나의 record로 효율적으로 그룹화시킬 수 있다.
서로 다른 데이터를 효과적으로 연결
- 위에서 만들었던 'customer' table과 연결하기 위한 또 다른 table 'orders'를 만들어보자.
CREATE TABLE orders(
id INT NOT NULL AUTO_INCREMENT,
customer_id INT NOT NULL,
product_id INT NOT NULL,
quantuty INT NOT NULL,
price INT NOT NULL,
PRIMARY KEY (id),
FOREIGN KEY (customer_id) REFERNCES customers (id),
FOREIGN KEY (product_id) REFERENCES products (id)
);
- 'orders' table은 customer_id를 'customers' table의 id에서 reference한다는 것을 알 수 있다.
- 이렇게 간단한 명령어 만으로 다른 table의 데이터를 끌어다 참조하여 두 table을 연결시킬 수 있다.
효율적인 query
- 특정 고객이 주문한 상품 목록을 보고싶다면 다음과 같이 SQL을 사용하면 된다.
orders.id,
orders.customer_id,
orders.product_id,
orders.quantity,
orders.price,
products.name
From oders
INNER JOIN products
ON orders.product_id=products.id
WHERE orders.customer_id=1;
Type of RDB
- MySQL
- Amazon Aurora
- PostgreSQL
- Microsoft SQL Server
- Oracale Database
- MariaDB
- SQL Server
참조




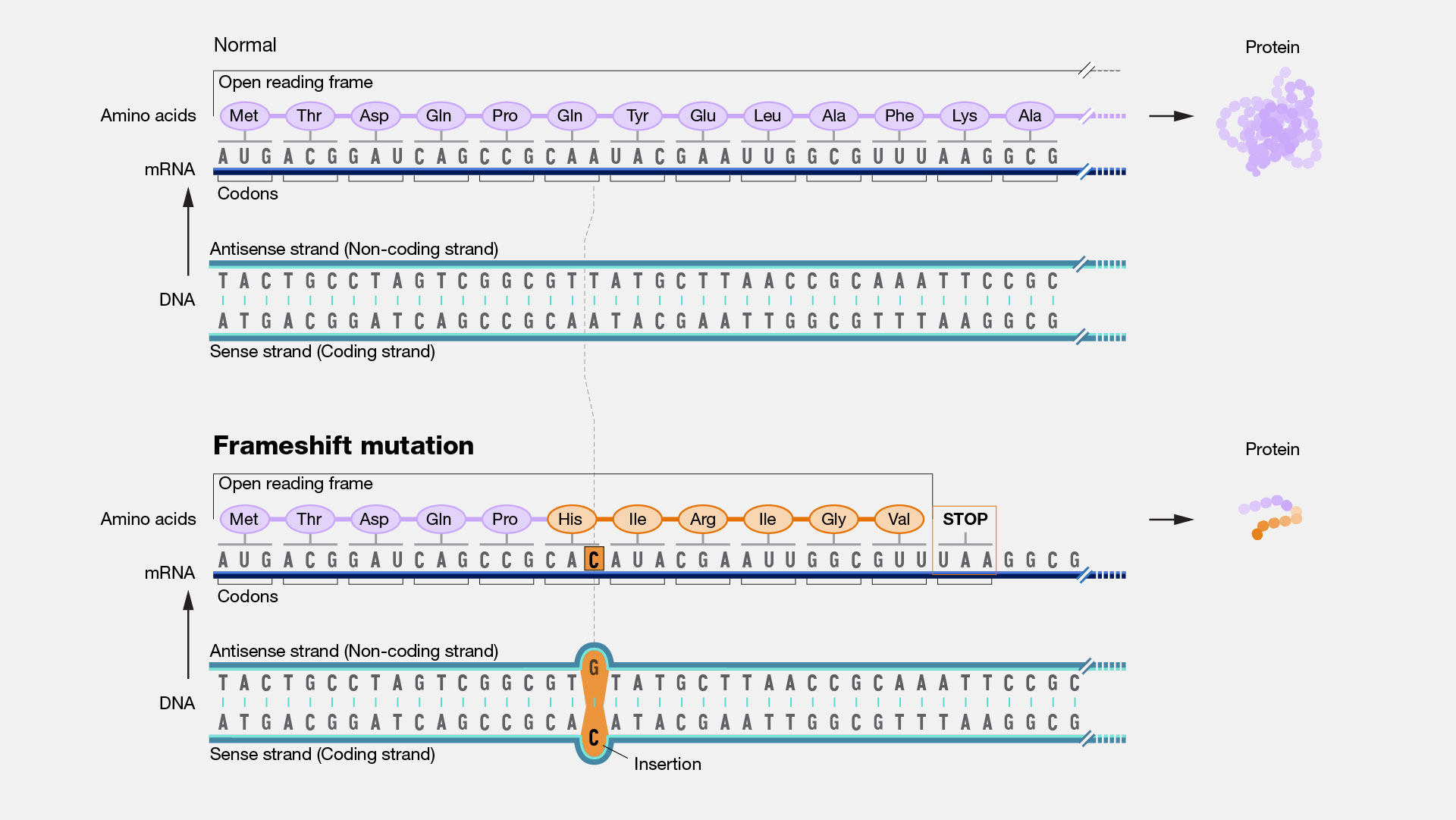
 1. snRNPs 5개 혹은 ribonucleoproteins 접근
2. intron에 붙음
3. intron의 양 끝(5', 4')이 모이면서 고리를 생성
4. intron이 제거됨
- intron은 이후 다른 과정에 참여함
6. snRNPs는 다시 재활용됨
1. snRNPs 5개 혹은 ribonucleoproteins 접근
2. intron에 붙음
3. intron의 양 끝(5', 4')이 모이면서 고리를 생성
4. intron이 제거됨
- intron은 이후 다른 과정에 참여함
6. snRNPs는 다시 재활용됨
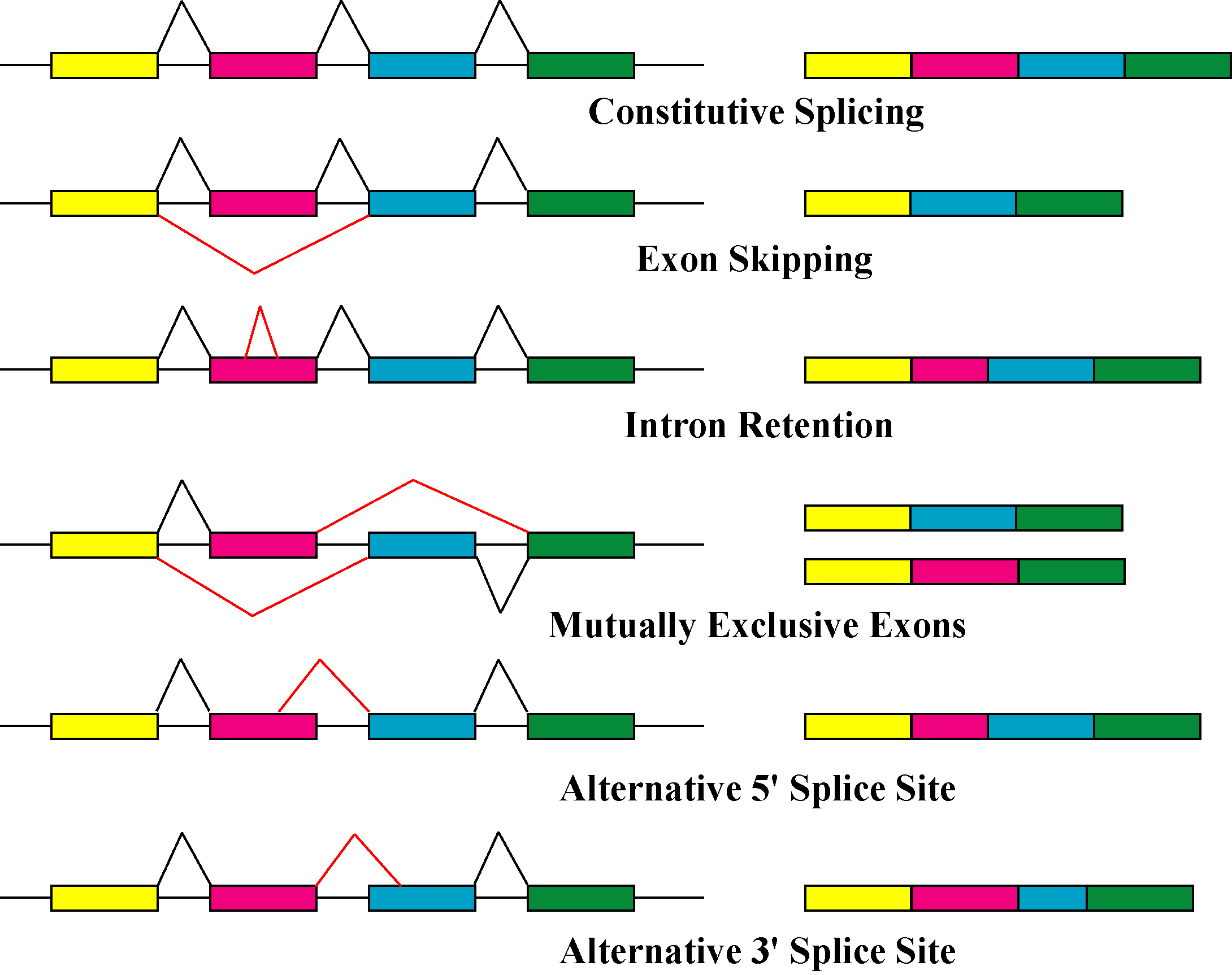 ## Exon Skipping (Cassette Exon)
- 특정 exon을 건너 뛰어 버린다.
## Intron Retention (Retained Intron)
- intron이 중간에 그대로 끼어있음
## Mutually Exclusive Exon
- 특정한 exon에서 여러 돌연변이가 발생해 다양한 종류의 돌연변이를 야기한다.
## Alternative 5' Donor Site
- upstream exon의 중간부분이 연결된다.
## Alternative 3' Acceptor Sites
- downsteram exon의 중간부분이 연결된다.
## Exon Skipping (Cassette Exon)
- 특정 exon을 건너 뛰어 버린다.
## Intron Retention (Retained Intron)
- intron이 중간에 그대로 끼어있음
## Mutually Exclusive Exon
- 특정한 exon에서 여러 돌연변이가 발생해 다양한 종류의 돌연변이를 야기한다.
## Alternative 5' Donor Site
- upstream exon의 중간부분이 연결된다.
## Alternative 3' Acceptor Sites
- downsteram exon의 중간부분이 연결된다.


 - 연산으로는 다음과 같이 나타낼 수 있다.
$$\huge f(x) * g(x) = h(x)$$
*f = filter, g= feature map, h= output*
# Deconvolution
- Deconvolution은 Convolution의 반대과정이다.
- 연산으로는 다음과 같이 나타낼 수 있다.
$$\huge f(x) * g(x) = h(x)$$
*f = filter, g= feature map, h= output*
# Deconvolution
- Deconvolution은 Convolution의 반대과정이다.